Penapisan maya atau skrining virtual (Bahasa Inggris: virtual screening, disingkat VS) adalah teknik komputasi yang digunakan dalam penemuan obat untuk mencari pustaka molekul kecil guna mengidentifikasi struktur yang paling mungkin berikatan dengan target obat, biasanya reseptor protein atau enzim.[1][2]
Penapisan maya telah didefinisikan sebagai "mengevaluasi pustaka senyawa yang sangat besar secara otomatis" menggunakan program komputer.[3] Seperti yang disarankan oleh definisi ini, VS sebagian besar merupakan permainan angka yang berfokus pada bagaimana ruang kimia yang sangat besar dari lebih dari 1060 senyawa yang mungkin[4] dapat disaring menjadi jumlah yang dapat dikelola yang dapat disintesis, dibeli, dan diuji. Meskipun pencarian seluruh ruang kimia mungkin merupakan masalah yang menarik secara teoritis, skenario VS yang lebih praktis berfokus pada perancangan dan pengoptimalan pustaka kombinatorial yang ditargetkan dan memperkaya pustaka senyawa yang tersedia dari repositori senyawa internal atau penawaran vendor. Seiring meningkatnya akurasi metode, penapisan maya telah menjadi bagian integral dari proses penemuan obat.[5][6] Penapisan maya dapat digunakan untuk memilih senyawa pangkalan data internal untuk penapisan, memilih senyawa yang dapat dibeli secara eksternal, dan untuk memilih senyawa mana yang harus disintesis selanjutnya.
Metode
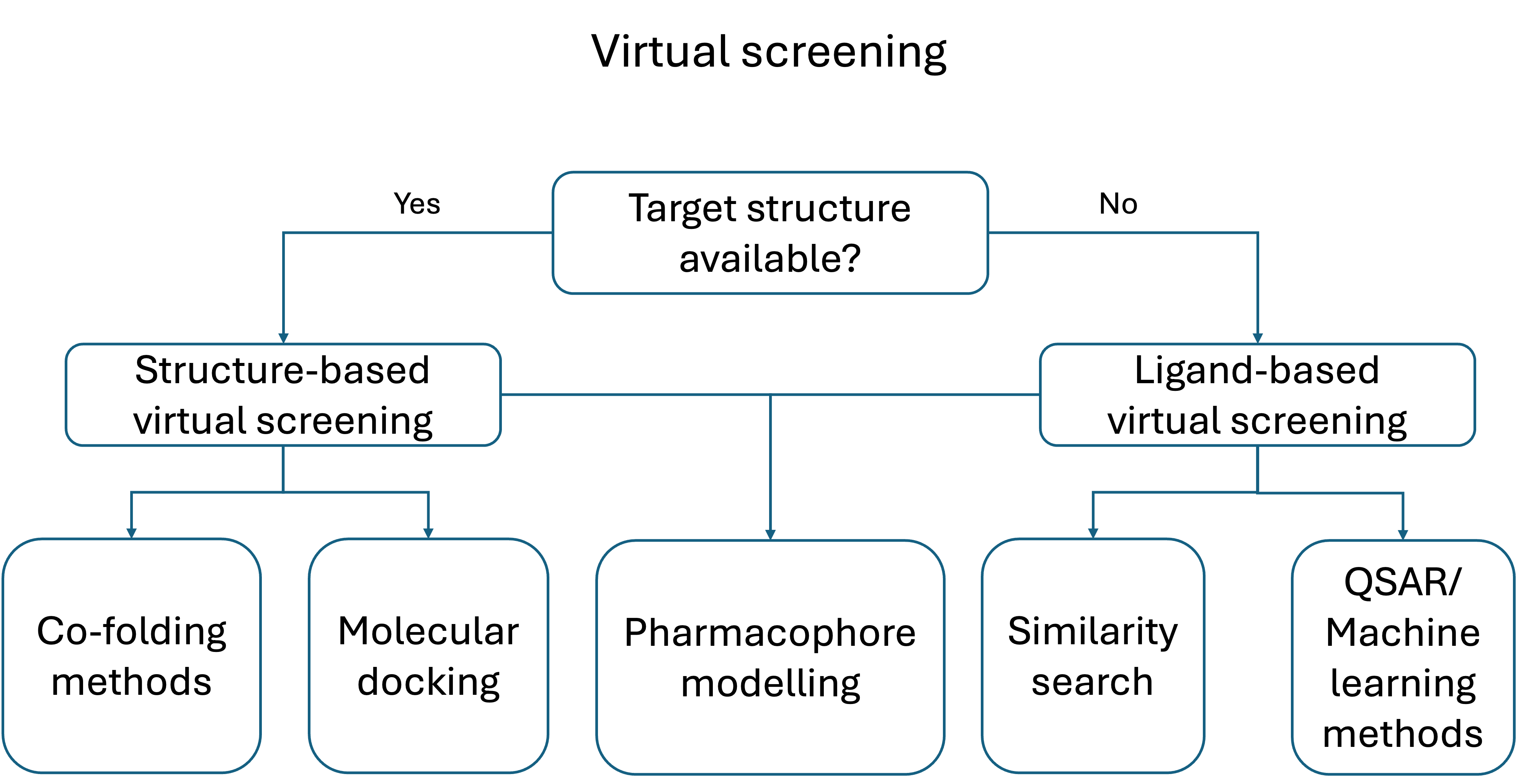
suntingTerdapat dua kategori besar teknik penapisan: berbasis ligan dan berbasis struktur.[7]
Metode berbasis ligan
suntingDengan adanya sekumpulan ligan yang beragam secara struktural yang mengikat reseptor, model reseptor dapat dibangun dengan memanfaatkan informasi kolektif yang terkandung dalam kumpulan ligan tersebut. Berbagai teknik komputasi mengeksplorasi kesamaan struktural, elektronik, bentuk molekuler, dan fisikokimia dari berbagai ligan yang dapat mengimplikasikan cara kerjanya terhadap reseptor molekuler atau lini sel tertentu.[8] Ligan kandidat kemudian dapat dibandingkan dengan model farmakofor untuk menentukan apakah ligan tersebut kompatibel dengannya dan oleh karena itu kemungkinan akan mengikat.[9] Berbagai metode analisis kesamaan kimia 2D[10] telah digunakan untuk memindai pangkalan data untuk menemukan ligan aktif. Pendekatan populer lainnya yang digunakan dalam penapisan maya berbasis ligan terdiri dari pencarian molekul dengan bentuk yang mirip dengan senyawa aktif yang diketahui, karena molekul tersebut akan sesuai dengan situs pengikatan target dan karenanya kemungkinan besar akan mengikat target. Terdapat sejumlah aplikasi prospektif dari kelas teknik ini dalam literatur.[11][12][13] Ekstensi farmakoforik dari metode 3D ini juga tersedia secara gratis sebagai server web.[14][15] Penapisan maya berbasis bentuk juga telah mendapatkan popularitas yang signifikan.[16]
Metode berbasis struktur
suntingPendekatan penapisan maya berbasis struktur mencakup berbagai teknik komputasi yang mempertimbangkan struktur reseptor yang merupakan target molekuler dari ligan aktif yang diteliti. Beberapa teknik ini termasuk penambatan molekuler, pemodelan farmakofor berbasis struktur, dan simulasi dinamika molekuler.[17][18][8] Penambatan molekuler adalah teknik berbasis struktur yang paling banyak digunakan, dan menerapkan fungsi penilaian untuk memperkirakan kesesuaian setiap ligan terhadap situs pengikatan reseptor makromolekuler, membantu memilih ligan dengan afinitas tertinggi.[19][20][21] Saat ini, terdapat beberapa server web yang berorientasi pada penapisan maya prospektif.[22][23]
Metode hibrida
suntingMetode hibrida yang bergantung pada kesamaan struktur dan ligan juga dikembangkan untuk mengatasi keterbatasan pendekatan VLS tradisional. Metodologi ini memanfaatkan informasi pengikatan ligan berbasis evolusi untuk memprediksi pengikat molekul kecil[24][25] dan dapat menggunakan kesamaan struktur global dan kesamaan kantong.[24] Pendekatan berbasis kesamaan struktur global menggunakan struktur eksperimental atau model protein yang diprediksi untuk menemukan kesamaan struktural dengan protein dalam pustaka templat holo PDB. Setelah mendeteksi kemiripan struktural yang signifikan, metrik koefisien Tanimoto berbasis sidik jari 2D diterapkan untuk menyaring molekul kecil yang mirip dengan ligan yang diekstrak dari templat holo PDB terpilih.[26][27] Prediksi dari metode ini telah dinilai secara eksperimental dan menunjukkan pengayaan yang baik dalam mengidentifikasi molekul kecil aktif.
Metode yang disebutkan di atas bergantung pada kemiripan struktural global dan tidak mampu secara priori memilih situs pengikatan ligan tertentu dalam protein yang diminati. Lebih lanjut, karena metode tersebut bergantung pada penilaian kemiripan 2D untuk ligan, metode tersebut tidak mampu mengenali kemiripan stereokimia molekul kecil yang secara substansial berbeda tetapi menunjukkan kemiripan bentuk geometris. Untuk mengatasi masalah ini, pendekatan baru yang berpusat pada kantong (PoLi) yang mampu menargetkan kantong pengikatan spesifik dalam templat holo-protein, dikembangkan dan dinilai secara eksperimental.
Infrastruktur komputasi
suntingPerhitungan interaksi berpasangan antar atom, yang merupakan prasyarat untuk pengoperasian banyak program penyaringan virtual, berskala , di mana N adalah jumlah atom dalam sistem. Karena penskalaan kuadratik, biaya komputasi meningkat dengan cepat.

Pendekatan berbasis ligan
suntingMetode berbasis ligan biasanya hanya membutuhkan sebagian kecil detik untuk satu operasi perbandingan struktur. Terkadang satu CPU sudah cukup untuk melakukan penyaringan besar dalam beberapa jam. Namun, beberapa perbandingan dapat dilakukan secara paralel untuk mempercepat pemrosesan pangkalan data senyawa yang besar.
Pendekatan berbasis struktur
suntingUkuran tugas membutuhkan infrastruktur komputasi paralel, seperti klaster sistem Linux, yang menjalankan prosesor antrian batch untuk menangani pekerjaan, seperti Sun Grid Engine atau Torque PBS.
Diperlukan cara untuk menangani input dari pustaka senyawa yang besar. Hal ini membutuhkan semacam pangkalan data senyawa yang dapat diakses oleh klaster paralel, yang mengirimkan senyawa secara paralel ke berbagai node komputasi. Mesin pangkalan data komersial mungkin terlalu lambat, dan mesin pengindeksan berkecepatan tinggi, seperti Berkeley DB, mungkin merupakan pilihan yang lebih baik. Selain itu, menjalankan satu perbandingan per pekerjaan mungkin tidak efisien, karena waktu pemanasan node klaster dapat dengan mudah melebihi jumlah pekerjaan yang bermanfaat. Untuk mengatasi hal ini, perlu untuk memproses kumpulan senyawa dalam setiap pekerjaan klaster, menggabungkan hasilnya ke dalam semacam log berkas. Proses sekunder, untuk menambang log berkas dan mengekstrak kandidat dengan skor tinggi, kemudian dapat dijalankan setelah seluruh percobaan selesai dijalankan.
Akurasi
suntingTujuan dari penapisan maya adalah untuk mengidentifikasi molekul dengan struktur kimia baru yang mengikat target makromolekuler yang diminati. Dengan demikian, keberhasilan penapisan maya didefinisikan dalam hal menemukan kerangka kerja baru yang menarik daripada jumlah total keberhasilan. Oleh karena itu, interpretasi akurasi penapisan maya harus dipertimbangkan dengan hati-hati. Tingkat keberhasilan yang rendah untuk kerangka molekul yang menarik jelas lebih disukai daripada tingkat keberhasilan yang tinggi untuk kerangka molekul yang sudah dikenal.
Sebagian besar pengujian studi penapisan maya dalam literatur bersifat retrospektif. Dalam studi ini, kinerja teknik VS diukur berdasarkan kemampuannya untuk mengambil sejumlah kecil molekul yang sebelumnya dikenal dengan afinitas terhadap target yang diminati (molekul aktif atau hanya aktif) dari pustaka yang mengandung proporsi molekul tidak aktif atau umpan yang jauh lebih tinggi. Ada beberapa cara berbeda untuk memilih umpan dengan mencocokkan sifat-sifat molekul aktif yang sesuai[28] dan baru-baru ini umpan juga dipilih dengan cara yang tidak sesuai sifatnya. Dampak sebenarnya dari pemilihan umpan, baik untuk tujuan pelatihan maupun pengujian, juga telah dibahas.[29][30]
Sebaliknya, dalam aplikasi prospektif penapisan maya, hasil keberhasilan tersebut tunduk pada konfirmasi eksperimental (misalnya pengukuran IC50). Terdapat konsensus bahwa tolok ukur retrospektif bukanlah prediktor yang baik untuk kinerja prospektif dan akibatnya hanya studi prospektif yang merupakan bukti konklusif tentang kesesuaian suatu teknik untuk target tertentu.[31][32][33][34][35]
Penggunaan untuk penemuan obat
suntingPenapisan maya merupakan penggunaan yang sangat berguna dalam mengidentifikasi molekul-molekul potensial sebagai awal dari kimia medisinal. Seiring dengan semakin pentingnya dan substansialnya pendekatan penapisan maya dalam industri kimia medisinal, pendekatan ini mengalami peningkatan yang pesat.[36]
Metode berbasis ligan
suntingMetode berbasis ligan diterapkan ketika struktur tiga dimensi reseptor target tidak diketahui. Alih-alih memodelkan interaksi ligan-reseptor secara langsung, pendekatan ini bergantung pada sifat struktural dan fisikokimia ligan aktif yang diketahui untuk memprediksi bagaimana senyawa baru dapat berikatan.[6]
Model farmakofor
suntingFarmakofor didefinisikan sebagai kumpulan fitur sterik dan elektronik yang diperlukan untuk memastikan interaksi supramolekuler optimal dengan target biologis dan untuk memicu (atau memblokir) respons biologisnya. Fitur-fitur ini biasanya mencakup donor dan akseptor ikatan hidrogen, daerah hidrofobik, cincin aromatik, gugus bermuatan, dan fungsi pengikat logam.[6][37]
Pemodelan farmakofor umumnya diterapkan dalam rancangan obat berbasis ligan ketika struktur tiga dimensi protein target tidak diketahui atau ketika beberapa ligan aktif tersedia.
Alur kerja umum melibatkan beberapa langkah:
- Pemilihan senyawa aktif: Seperangkat ligan aktif biologis yang representatif dan beragam secara struktural dipilih. Keragaman struktural penting untuk memastikan bahwa model yang dihasilkan menangkap fitur interaksi penting daripada karakteristik spesifik senyawa.
- Pembuatan konformer: Karena ligan bersifat fleksibel, beberapa konformasi berenergi rendah (konformer) dihasilkan untuk setiap molekul. Langkah ini bertujuan untuk mendekati konformasi bioaktif, yang biasanya tidak diketahui dalam pendekatan berbasis ligan.
- Penyelarasan dan superposisi molekuler: Konformer yang dihasilkan ditumpangkan untuk mengidentifikasi susunan spasial umum dari fitur kimia utama. Dengan menyelaraskan elemen farmakoforik bersama di seluruh ligan aktif, hipotesis farmakofor dibangun.
Tidak seperti pendekatan yang bergantung pada satu struktur referensi, pemodelan farmakofor mengintegrasikan informasi dari beberapa senyawa aktif. Ini umumnya meningkatkan kekokohan dan kinerja prediktif, terutama ketika berurusan dengan ligan yang beragam secara kimia.
Namun, karena banyak kemungkinan penyelarasan dan kombinasi fitur, pemodelan farmakofor biasanya tidak menghasilkan satu solusi unik. Oleh karena itu, hipotesis yang dihasilkan harus divalidasi menggunakan set uji eksternal atau data eksperimental.
Penapisan maya berbasis bentuk
suntingPendekatan kemiripan molekuler berbasis bentuk telah ditetapkan sebagai teknik penapisan maya yang penting dan populer. Saat ini, platform penyaringan yang sangat optimal ROCS (Rapid Overlay of Chemical Structures) dianggap sebagai standar industri de facto untuk penapisan maya berbasis bentuk dan berpusat pada ligan.[38][39][40] Platform ini menggunakan fungsi Gaussian untuk mendefinisikan volume molekuler molekul organik kecil. Pemilihan konformasi kueri kurang penting, sehingga penyaringan berbasis bentuk ideal untuk pemodelan berbasis ligan: Karena ketersediaan konformasi bioaktif untuk kueri bukanlah faktor pembatas untuk penyaringan, yang lebih menentukan kinerja penyaringan adalah pemilihan senyawa kueri.[16] Metode kesamaan molekul berbasis bentuk lainnya seperti Autodock-SS juga telah dikembangkan.[41]
Penapisan maya berbasis medan
suntingMetode penapisan maya berbasis medan memperluas pendekatan kesamaan berbasis bentuk dengan mempertimbangkan tidak hanya bentuk molekul tetapi juga medan interaksi fisikokimia yang mengatur pengenalan ligan-reseptor.
Alih-alih hanya berfokus pada tumpang tindih struktural, metode ini membandingkan potensi interaksi molekul, seperti:
- Medan elektrostatik
- Medan hidrofobik
- Medan sterik
- Potensi ikatan hidrogen
Dengan mengevaluasi sifat-sifat ini dalam ruang tiga dimensi, pendekatan berbasis medan bertujuan untuk menangkap pola interaksi mendasar yang bertanggung jawab atas aktivitas biologis, sementara sebagian besar tetap independen dari kerangka kimia spesifik molekul yang diteliti.
Dibandingkan dengan metode yang murni berbasis bentuk, penyaringan berbasis medan dapat memberikan deskripsi kesamaan molekul yang lebih bernuansa, terutama ketika senyawa yang berbeda secara struktural memiliki profil interaksi yang serupa.[42][43]
Hubungan struktur-aktivitas kuantitatif
suntingModel hubungan kuantitatif struktur–aktivitas (QSAR) adalah model prediktif yang menghubungkan deskriptor molekuler dengan aktivitas biologis menggunakan kumpulan data senyawa aktif dan tidak aktif yang diketahui. Berbeda dengan analisis hubungan kualitatif struktur–aktivitas (SAR), yang mengidentifikasi tren dalam kelas struktural, QSAR memberikan hubungan matematis kuantitatif antara sifat molekuler dan respons biologis yang terukur.[6][44]
Metode QSAR tradisional
suntingPendekatan QSAR tradisional biasanya bergantung pada deskriptor molekuler yang telah ditentukan sebelumnya; seperti sifat fisikokimia, indeks topologi, atau parameter elektronik; dan menerapkan teknik pemodelan statistik termasuk:
- Kuadrat terkecil biasa (OLS)
- Regresi linier berganda (MLR)
- Kuadrat terkecil parsial (PLS)
Metode-metode ini mengasumsikan hubungan linier antara deskriptor dan aktivitas biologis. Model QSAR banyak digunakan untuk memprioritaskan senyawa dalam penemuan dan optimasi kandidat obat.
Pembelajaran mesin dalam QSAR
suntingPendekatan pembelajaran mesin (ML) dapat dilihat sebagai perluasan metodologi QSAR. Seperti model QSAR klasik, metode berbasis ML menggunakan deskriptor molekuler atau representasi struktural (seperti sidik jari molekuler,[45] grafik) sebagai fitur masukan. Namun, mereka menggunakan algoritma yang lebih fleksibel yang mampu memodelkan hubungan nonlinier dan kompleks antara struktur molekuler dan aktivitas biologis.
Dalam pembelajaran terbimbing, model dilatih pada kumpulan data yang terdiri dari senyawa aktif dan tidak aktif yang diketahui (atau senyawa dengan nilai aktivitas terukur). Model yang terlatih kemudian digunakan untuk memprediksi aktivitas atau probabilitas aktivitas untuk senyawa baru.
Algoritma pembelajaran mesin umum yang diterapkan dalam penyaringan virtual meliputi:[46][47][48]
- Pohon keputusan
- Mesin vektor pendukung (SVM)
- Random forest
- k-Tetangga terdekat (k-NN)
- Jaringan saraf tiruan
Model-model ini biasanya menghasilkan:
- Nilai aktivitas yang diprediksi (regresi), atau
- Probabilitas bahwa suatu senyawa aktif (klasifikasi), yang kemudian dapat digunakan untuk pemeringkatan dalam penyaringan virtual.
Metode berbasis struktur
suntingMetode berbasis struktur bergantung pada struktur tiga dimensi target biologis, yang biasanya diperoleh dari kristalografi sinar-X, spektroskopi NMR, atau mikroskopi krio-elektron. Tidak seperti pendekatan berbasis ligan, metode ini secara eksplisit memodelkan interaksi antara ligan dan situs pengikatan reseptor.
Model farmakofor
suntingDalam pemodelan farmakofor berbasis struktur, fitur farmakoforik diturunkan langsung dari struktur tiga dimensi situs pengikatan target, bukan dari sekumpulan ligan aktif yang diketahui.
Fitur interaksi utama; seperti donor dan akseptor ikatan hidrogen, daerah hidrofobik, residu bermuatan, dan situs pengikatan logam; diidentifikasi berdasarkan susunan spasial residu asam amino di dalam kantung pengikatan.[49][50] Jika struktur kompleks protein-ligan tersedia, fitur farmakoforik juga dapat diekstrak dari interaksi ligan-reseptor yang diamati.[51]
Model farmakofor yang dihasilkan mewakili pola interaksi penting yang diperlukan untuk pengikatan dan dapat digunakan untuk menyaring pustaka senyawa.
Penambatan molekuler
suntingPenambatan molekuler bertujuan untuk memprediksi mode pengikatan (pose), dan dalam beberapa kasus afinitas pengikatan ligan di dalam situs aktif protein target.[52]
Penambatan melibatkan dua komponen utama:
- Algoritma pencarian: mengeksplorasi kemungkinan orientasi dan konformasi ligan di dalam kantung pengikatan.
- Fungsi penilaian: memperkirakan kekuatan interaksi ligan-reseptor dan memberi peringkat pose yang diprediksi sesuai dengan itu.
Tujuannya adalah untuk mengidentifikasi pose pengikatan yang paling mungkin untuk ligan tertentu dan untuk memprioritaskan senyawa berdasarkan afinitas pengikatan yang diprediksi.[6][53]
Metode co-folding
suntingMetode co-folding memprediksi struktur kompleks protein-ligan dengan memodelkan reseptor dan ligan secara bersamaan, memungkinkan adaptasi konformasi selama pengikatan. Berbeda dengan penambatan klasik, yang biasanya memperlakukan reseptor sebagai kaku atau semi-fleksibel, pendekatan co-folding bertujuan untuk menangkap efek induced-fit secara lebih eksplisit.
Kerangka kerja berbasis deep learning terbaru seperti Boltz-2[54] mencontohkan strategi ini dengan memprediksi struktur kompleks terikat langsung dari informasi urutan dan ligan. Meskipun pendekatan tersebut secara komputasi lebih menuntut daripada penambatan tradisional, pendekatan ini menjanjikan untuk menyempurnakan posisi pengikatan, meningkatkan akurasi struktural, dan meningkatkan alur kerja penapisan maya berbasis struktur, khususnya untuk target yang fleksibel atau menantang.
Lihat juga
sunting- Komputasi grid
- Penapisan lewatan tinggi
- Penambatan (molekuler)
- Penapisan retro
- Fungsi penilaian
- Penapisan skala ultra besar
- ZINC
Referensi
sunting- ^ Rester U (July 2008). "From virtuality to reality - Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective". Current Opinion in Drug Discovery & Development. 11 (4): 559–68. PMID 18600572.
- ^ Rollinger JM, Stuppner H, Langer T (2008). "Virtual screening for the discovery of bioactive natural products". Natural Compounds as Drugs Volume I. Progress in Drug Research. Fortschritte der Arzneimittelforschung. Progrès des Recherches Pharmaceutiques. Vol. 65. hlm. 211, 213–49. doi:10.1007/978-3-7643-8117-2_6. ISBN 978-3-7643-8098-4. PMC 7124045. PMID 18084917.
- ^ Walters WP, Stahl MT, Murcko MA (1998). "Virtual screening – an overview". Drug Discov. Today. 3 (4): 160–178. doi:10.1016/S1359-6446(97)01163-X.
- ^ Bohacek RS, McMartin C, Guida WC (1996). "The art and practice of structure-based drug design: a molecular modeling perspective". Med. Res. Rev. 16 (1): 3–50. doi:10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. PMID 8788213.
- ^ McGregor MJ, Luo Z, Jiang X (June 11, 2007). "Chapter 3: Virtual screening in drug discovery". Dalam Huang Z (ed.). Drug Discovery Research. New Frontiers in the Post-Genomic Era. Wiley-VCH: Weinheim, Germany. hlm. 63–88. ISBN 978-0-471-67200-5.
- ^ a b c d e Gillet V (2013). "Ligand-Based and Structure-Based Virtual Screening" (PDF). The University of Sheffield.
- ^ McInnes C (October 2007). "Virtual screening strategies in drug discovery". Current Opinion in Chemical Biology. 11 (5): 494–502. doi:10.1016/j.cbpa.2007.08.033. PMID 17936059.
- ^ a b Santana K, do Nascimento LD, Lima e Lima A, Damasceno V, Nahum C, Braga RC, Lameira J (2021-04-29). "Applications of Virtual Screening in Bioprospecting: Facts, Shifts, and Perspectives to Explore the Chemo-Structural Diversity of Natural Products". Frontiers in Chemistry. 9 662688. Bibcode:2021FrCh....9..155S. doi:10.3389/fchem.2021.662688. ISSN 2296-2646. PMC 8117418. PMID 33996755.
- ^ Sun H (2008). "Pharmacophore-based virtual screening". Current Medicinal Chemistry. 15 (10): 1018–24. doi:10.2174/092986708784049630. PMID 18393859.
- ^ Willet P, Barnard JM, Downs GM (1998). "Chemical similarity searching". Journal of Chemical Information and Computer Sciences. 38 (6): 983–996. CiteSeerX 10.1.1.453.1788. doi:10.1021/ci9800211.
- ^ Rush TS, Grant JA, Mosyak L, Nicholls A (March 2005). "A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction". Journal of Medicinal Chemistry. 48 (5): 1489–95. CiteSeerX 10.1.1.455.4728. doi:10.1021/jm040163o. PMID 15743191.
- ^ Ballester PJ, Westwood I, Laurieri N, Sim E, Richards WG (February 2010). "Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases". Journal of the Royal Society, Interface. 7 (43): 335–42. doi:10.1098/rsif.2009.0170. PMC 2842611. PMID 19586957.
- ^ Kumar A, Zhang KY (2018). "Advances in the Development of Shape Similarity Methods and Their Application in Drug Discovery". Frontiers in Chemistry (dalam bahasa English). 6 315. Bibcode:2018FrCh....6..315K. doi:10.3389/fchem.2018.00315. PMC 6068280. PMID 30090808. Pemeliharaan CS1: Bahasa yang tidak diketahui (link)
- ^ Li H, Leung KS, Wong MH, Ballester PJ (July 2016). "USR-VS: a web server for large-scale prospective virtual screening using ultrafast shape recognition techniques". Nucleic Acids Research (dalam bahasa Inggris). 44 (W1): W436–41. doi:10.1093/nar/gkw320. PMC 4987897. PMID 27106057.
- ^ Sperandio O, Petitjean M, Tuffery P (July 2009). "wwLigCSRre: a 3D ligand-based server for hit identification and optimization". Nucleic Acids Research. 37 (Web Server issue): W504–9. doi:10.1093/nar/gkp324. PMC 2703967. PMID 19429687.
- ^ a b Kirchmair J, Distinto S, Markt P, Schuster D, Spitzer GM, Liedl KR, Wolber G (2009). "How To Optimize Shape-Based Virtual Screening: Choosing the Right Query and Including Chemical Information". Journal of Chemical Information and Modeling. 49 (3): 678–692. doi:10.1021/ci8004226. PMID 19434901.
- ^ Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, Kalid O (July 2014). "Structure-based virtual screening approach for discovery of covalently bound ligands". Journal of Chemical Information and Modeling. 54 (7): 1941–50. doi:10.1021/ci500175r. PMID 24932913.
- ^ Maia EH, Assis LC, de Oliveira TA, da Silva AM, Taranto AG (2020-04-28). "Structure-Based Virtual Screening: From Classical to Artificial Intelligence". Frontiers in Chemistry. 8 343. Bibcode:2020FrCh....8..343M. doi:10.3389/fchem.2020.00343. PMC 7200080. PMID 32411671.
- ^ Kroemer RT (August 2007). "Structure-based drug design: docking and scoring". Current Protein & Peptide Science. 8 (4): 312–28. CiteSeerX 10.1.1.225.959. doi:10.2174/138920307781369382. PMID 17696866.
- ^ Cavasotto CN, Orry AJ (2007). "Ligand docking and structure-based virtual screening in drug discovery". Current Topics in Medicinal Chemistry. 7 (10): 1006–14. doi:10.2174/156802607780906753. PMID 17508934.
- ^ Kooistra AJ, Vischer HF, McNaught-Flores D, Leurs R, de Esch IJ, de Graaf C (June 2016). "Function-specific virtual screening for GPCR ligands using a combined scoring method". Scientific Reports. 6 28288. Bibcode:2016NatSR...628288K. doi:10.1038/srep28288. PMC 4919634. PMID 27339552.
- ^ Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y (September 2009). "Automated docking screens: a feasibility study". Journal of Medicinal Chemistry. 52 (18): 5712–20. doi:10.1021/jm9006966. PMC 2745826. PMID 19719084.
- ^ Li H, Leung KS, Ballester PJ, Wong MH (2014-01-24). "istar: a web platform for large-scale protein-ligand docking". PLOS ONE. 9 (1) e85678. Bibcode:2014PLoSO...985678L. doi:10.1371/journal.pone.0085678. PMC 3901662. PMID 24475049.
- ^ a b Zhou H, Skolnick J (January 2013). "FINDSITE(comb): a threading/structure-based, proteomic-scale virtual ligand screening approach". Journal of Chemical Information and Modeling (dalam bahasa Inggris). 53 (1): 230–40. doi:10.1021/ci300510n. PMC 3557555. PMID 23240691.
- ^ Roy A, Skolnick J (February 2015). "LIGSIFT: an open-source tool for ligand structural alignment and virtual screening". Bioinformatics. 31 (4): 539–44. doi:10.1093/bioinformatics/btu692. PMC 4325547. PMID 25336501.
- ^ Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (January 2012). "ChEMBL: a large-scale bioactivity database for drug discovery". Nucleic Acids Research. 40 (Database issue): D1100–7. doi:10.1093/nar/gkr777. PMC 3245175. PMID 21948594.
- ^ Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (January 2006). "DrugBank: a comprehensive resource for in silico drug discovery and exploration". Nucleic Acids Research. 34 (Database issue): D668–72. doi:10.1093/nar/gkj067. PMC 1347430. PMID 16381955.
- ^ Réau M, Langenfeld F, Zagury JF, Lagarde N, Montes M (2018). "Decoys Selection in Benchmarking Datasets: Overview and Perspectives". Frontiers in Pharmacology. 9 11. doi:10.3389/fphar.2018.00011. PMC 5787549. PMID 29416509.
- ^ Ballester PJ (December 2019). "Selecting machine-learning scoring functions for structure-based virtual screening". Drug Discovery Today: Technologies. 32–33: 81–87. doi:10.1016/j.ddtec.2020.09.001. PMID 33386098. S2CID 224968364.
- ^ Li H, Sze KH, Lu G, Ballester PJ (2021). "Machine-learning scoring functions for structure-based virtual screening". WIREs Computational Molecular Science (dalam bahasa Inggris). 11 (1) e1478. doi:10.1002/wcms.1478. ISSN 1759-0884. S2CID 219089637.
- ^ Wallach I, Heifets A (2018). "Most Ligand-based classification benchmarks reward memorization rather than generalization". Journal of Chemical Information and Modeling. 58 (5): 916–932. arXiv:1706.06619. doi:10.1021/acs.jcim.7b00403. PMID 29698607. S2CID 195345933.
- ^ Irwin JJ (2008). "Community benchmarks for virtual screening". Journal of Computer-Aided Molecular Design. 22 (3–4): 193–9. Bibcode:2008JCAMD..22..193I. doi:10.1007/s10822-008-9189-4. PMID 18273555. S2CID 26260725.
- ^ Good AC, Oprea TI (2008). "Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection?". Journal of Computer-Aided Molecular Design. 22 (3–4): 169–78. Bibcode:2008JCAMD..22..169G. doi:10.1007/s10822-007-9167-2. PMID 18188508. S2CID 7738182.
- ^ Schneider G (April 2010). "Virtual screening: an endless staircase?". Nature Reviews. Drug Discovery. 9 (4): 273–6. doi:10.1038/nrd3139. PMID 20357802. S2CID 205477076.
- ^ Ballester PJ (January 2011). "Ultrafast shape recognition: method and applications". Future Medicinal Chemistry. 3 (1): 65–78. doi:10.4155/fmc.10.280. PMID 21428826.
- ^ Lavecchia A, Di Giovanni C (2013). "Virtual screening strategies in drug discovery: a critical review". Current Medicinal Chemistry. 20 (23): 2839–60. doi:10.2174/09298673113209990001. PMID 23651302.
- ^ Spitzer GM, Heiss M, Mangold M, Markt P, Kirchmair J, Wolber G, Liedl KR (2010). "One concept, three implementations of 3D pharmacophore-based virtual screening: distinct coverage of chemical search space". Journal of Chemical Information and Modeling. 50 (7): 1241–1247. doi:10.1021/ci100136b. PMID 20583761.
- ^ Grant JA, Gallard MA, Pickup BT (1996). "A fast method of molecular shape comparison: a simple application of a Gaussian description of molecular shape". Journal of Computational Chemistry. 17 (14): 1653–1666. doi:10.1002/(SICI)1096-987X(19961115)17:14<1653::AID-JCC7>3.0.CO;2-K.
- ^ Nicholls A, Grant JA (2005). "Molecular shape and electrostatics in the encoding of relevant chemical information". Journal of Computer-Aided Molecular Design. 19 (9–10): 661–686. Bibcode:2005JCAMD..19..661N. doi:10.1007/s10822-005-9019-x. PMID 16328855.
- ^ Rush TS, Grant JA, Mosyak L, Nicholls A (2005). "A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction". Journal of Medicinal Chemistry. 48 (5): 1489–1495. doi:10.1021/jm040163o. PMID 15743191.
- ^ Ni B, Wang H, Khalaf HK, Blay V, Houston DR (May 2024). "AutoDock-SS: AutoDock for Multiconformational Ligand-Based Virtual Screening". Journal of Chemical Information and Modeling. 64 (9): 3779–3789. doi:10.1021/acs.jcim.4c00136. PMC 11094722. PMID 38624083.
- ^ Cheeseright TJ, Mackey MD, Melville JL, Vinter JG (November 2008). "FieldScreen: virtual screening using molecular fields. Application to the DUD data set". Journal of Chemical Information and Modeling. 48 (11): 2108–2117. doi:10.1021/ci800110p. PMID 18991371.
- ^ Lang S, Slater MJ (May 2024). "Virtual Screening Strategies for Identifying Novel Chemotypes". Journal of Medicinal Chemistry. 67 (9): 6897–6898. doi:10.1021/acs.jmedchem.4c00906. PMID 38654500.
- ^ Neves BJ, Braga RC, Melo-Filho CC, Moreira-Filho JT, Muratov EN, Andrade CH (2018-11-13). "QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery". Frontiers in Pharmacology. 9 1275. doi:10.3389/fphar.2018.01275. PMC 6262347. PMID 30524275.
- ^ Luttens, Andreas; Cabeza de Vaca, Israel; Sparring, Leonard; Brea, José; Martínez, Antón Leandro; Kahlous, Nour Aldin; Radchenko, Dmytro S.; Moroz, Yurii S.; Loza, María Isabel; Norinder, Ulf; Carlsson, Jens (2025-03-13). "Rapid traversal of vast chemical space using machine learning-guided docking screens". Nature Computational Science (dalam bahasa Inggris). 5 (4): 301–312. doi:10.1038/s43588-025-00777-x. ISSN 2662-8457. PMC 12021657. PMID 40082701.
- ^ Alsenan S, Al-Turaiki I, Hafez A (December 2020). "A Recurrent Neural Network model to predict blood-brain barrier permeability". Computational Biology and Chemistry. 89 107377. doi:10.1016/j.compbiolchem.2020.107377. PMID 33010784.
- ^ Dimitri GM, Lió P (June 2017). "DrugClust: A machine learning approach for drugs side effects prediction". Computational Biology and Chemistry. 68: 204–210. doi:10.1016/j.compbiolchem.2017.03.008. PMID 28391063.
- ^ Shoombuatong W, Schaduangrat N, Pratiwi R, Nantasenamat C (June 2019). "THPep: A machine learning-based approach for predicting tumor homing peptides". Computational Biology and Chemistry. 80: 441–451. doi:10.1016/j.compbiolchem.2019.05.008. PMID 31151025.
- ^ Tran, Que-Huong; Nguyen, Quoc-Thai; Vo, Nguyen-Quynh-Huong; Mai, Tan Thanh; Tran, Thi-Thuy-Nga; Tran, Thanh-Dao; Le, Minh-Tri; Trinh, Dieu-Thuong Thi; Thai, Khac-Minh (2022-04-06). "Structure-based 3D-Pharmacophore modeling to discover novel interleukin 6 inhibitors: An in silico screening, molecular dynamics simulations and binding free energy calculations". PLOS ONE (dalam bahasa Inggris). 17 (4) e0266632. Bibcode:2022PLoSO..1766632T. doi:10.1371/journal.pone.0266632. ISSN 1932-6203. PMC 8986010. PMID 35385549.
- ^ Le, M.-T.; Mai, T.T.; Huynh, P.N.H.; Tran, T.-D.; Thai, K.-M.; Nguyen, Q.-T. (2020-12-01). "Structure-based discovery of interleukin-33 inhibitors: a pharmacophore modelling, molecular docking, and molecular dynamics simulation approach". SAR and QSAR in Environmental Research (dalam bahasa Inggris). 31 (12): 883–904. Bibcode:2020SQER...31..883L. doi:10.1080/1062936X.2020.1837239. ISSN 1062-936X. PMID 33191795.
- ^ Koes, D. R.; Camacho, C. J. (2012-07-01). "ZINCPharmer: pharmacophore search of the ZINC database". Nucleic Acids Research (dalam bahasa Inggris). 40 (W1): W409 – W414. doi:10.1093/nar/gks378. ISSN 0305-1048. PMC 3394271. PMID 22553363.
- ^ Bender, Brian J.; Gahbauer, Stefan; Luttens, Andreas; Lyu, Jiankun; Webb, Chase M.; Stein, Reed M.; Fink, Elissa A.; Balius, Trent E.; Carlsson, Jens; Irwin, John J.; Shoichet, Brian K. (2021). "A practical guide to large-scale docking". Nature Protocols (dalam bahasa Inggris). 16 (10): 4799–4832. doi:10.1038/s41596-021-00597-z. ISSN 1750-2799. PMC 8522653. PMID 34561691.
- ^ Pradeepkiran JA, Reddy PH (March 2019). "Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer's Disease". Cells. 8 (3): 260. doi:10.3390/cells8030260. PMC 6468864. PMID 30893872.
- ^ Passaro, Saro; Corso, Gabriele; Wohlwend, Jeremy; Reveiz, Mateo; Thaler, Stephan; Somnath, Vignesh Ram; Getz, Noah; Portnoi, Tally; Roy, Julien (2025-06-18). "Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction" (dalam bahasa Inggris). bioRxiv 10.1101/2025.06.14.659707.
Bacaan lanjutan
sunting- Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (May 2007). "Optimization of biaryl piperidine and 4-amino-2-biarylurea MCH1 receptor antagonists using QSAR modeling, classification techniques and virtual screening". Journal of Computer-Aided Molecular Design. 21 (5): 251–67. Bibcode:2007JCAMD..21..251M. doi:10.1007/s10822-007-9112-4. PMID 17377847. S2CID 19563229.
- Afantitis A, Melagraki G, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (February 2006). "Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques". Journal of Computer-Aided Molecular Design. 20 (2): 83–95. Bibcode:2006JCAMD..20...83A. CiteSeerX 10.1.1.716.8148. doi:10.1007/s10822-006-9038-2. PMID 16783600. S2CID 21523436.
- Eckert H, Bajorath J (March 2007). "Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches". Drug Discovery Today. 12 (5–6): 225–33. doi:10.1016/j.drudis.2007.01.011. PMID 17331887.
- Willett P (December 2006). "Similarity-based virtual screening using 2D fingerprints" (PDF). Drug Discovery Today (Submitted manuscript). 11 (23–24): 1046–53. doi:10.1016/j.drudis.2006.10.005. PMID 17129822.
- Fara DC, Oprea TI, Prossnitz ER, Bologa CG, Edwards BS, Sklar LA (2006). "Integration of virtual and physical screening". Drug Discovery Today: Technologies. 3 (4): 377–385. doi:10.1016/j.ddtec.2006.11.003. PMC 7105924. PMID 38620118.
- Muegge I, Oloffa S (2006). "Advances in virtual screening". Drug Discovery Today: Technologies. 3 (4): 405–411. doi:10.1016/j.ddtec.2006.12.002. PMC 7105922. PMID 38620182.
- Schneider G (April 2010). "Virtual screening: an endless staircase?". Nature Reviews. Drug Discovery. 9 (4): 273–6. doi:10.1038/nrd3139. PMID 20357802. S2CID 205477076.
Pranala luar
sunting- VLS3D – list of over 2000 databases, online and standalone in silico tools
